Publications - Dr. John Smith.

High Resolution multidimensional Biological NMR.
|
|---|
|
Grants:
BBSRC Structural Biology and Design Initiative.
Awarded Ł141,536, ran from Nov. 1997 - Nov. 2000.
Prof. J.B.Jackson, Dr. S.A. White & Dr. K.J.Smith
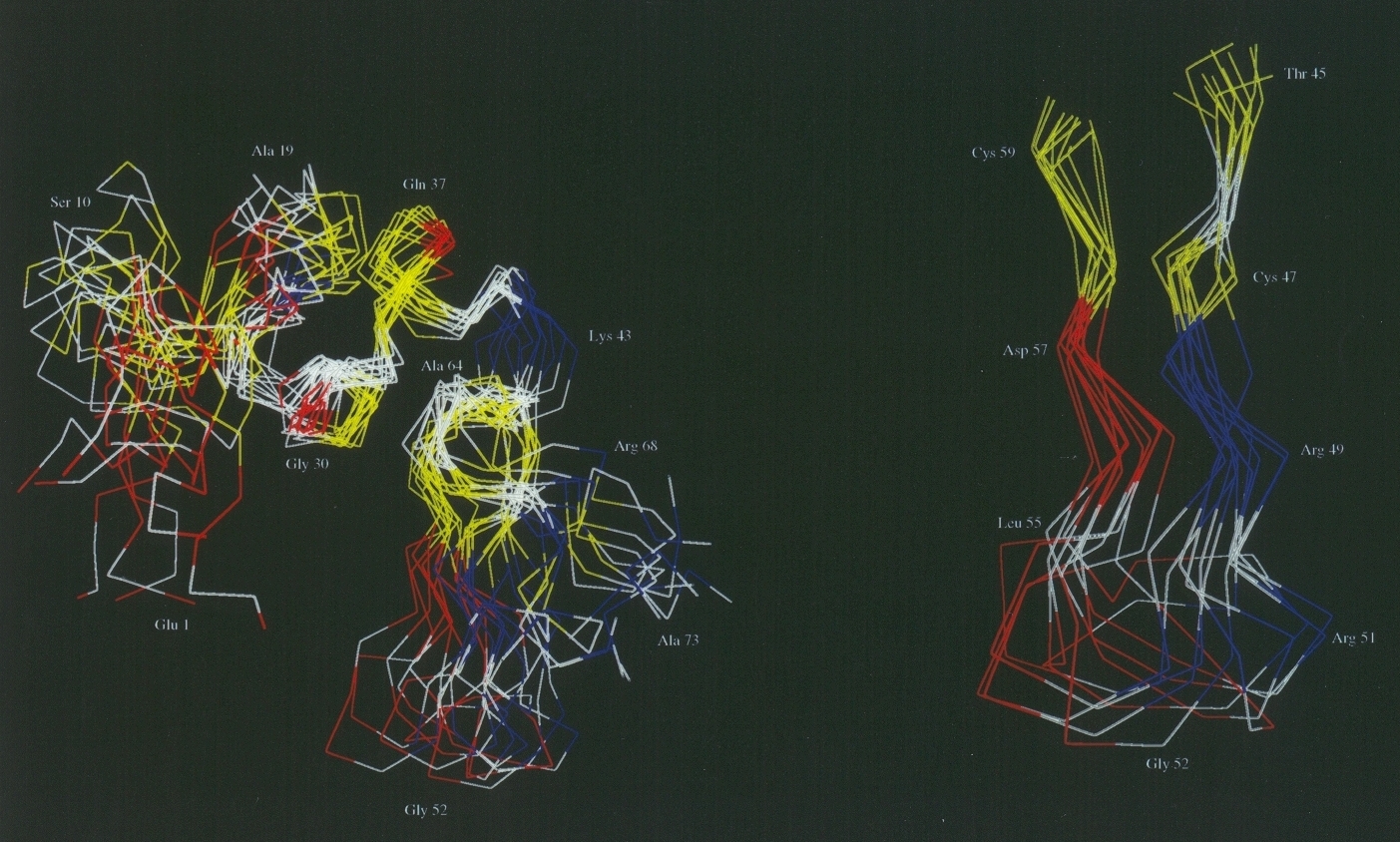
The high resolution structure of domain III of proton-translocating
nicotinamide nucleotide transhydrogenase.
|
|---|
|
with Dr. Mark Jeeves
see the BBSRC
Structural Biology and Design Initiative site and search the
research database
for Grant Reference: SBD07558.
Abstract:
Transhydrogenase pumps protons across a membrane. Domain III of the
protein probably contains the essential machinery that couples the
chemical reaction (hydride transfer between NAD(H) and NADP(H) to H+
translocation. We have expressed and purified domain III protein from
bacterial transhydrogenase in large quantities. The recombinant protein
has a very high affinity for NADP(H) and, together with the ND(H)-binding
domain I protein, can catalyse hydride transfer, even in the absence of
the membrane-intercalating domain II. Recombinant domain III protein of
transhydrogenase is soluble and stable; it has an excellent 1H- 1H 2D NMR
spectrum. The primary objective of this work is to solve the
high-resolution structure of the protein using NMR spectroscopy. We shall
also initiate crystallisation trials of the domain I:III complex with a
view to obtaining high resolution structure by X-ray diffraction.
Publications relating to this grant:
|
|
Publications:
|
|
Brear,K. Currey,J.D. Raines,S. & Smith,K.J.
(1988) Engineering in Medicine, 17, 4, 163-167.
Density and temperature effects on some of the mechanical
properties of cancellous bone.
|
|---|
|
Local backup of Full text PDF
This project was done at
York University.
the journal "Engineering in Medicine" is now defunct.
|
|
Keane,A.M. Smith.K.J.. & Trayer,I.P.
(1990) Biochem.Soc.Trans. 18, 264-266.
Short Report: The use of peptide mimetics and proton magnetic
resonance to define actin binding sites on the myosin head.
|
|---|
|
Local backup of Full text PDF
get this article from the
Biochemical Society Transactions site
|
|
Trayer,I.P., Keane,A.M., Murad,Z., Ruegg,J. & Smith,K.J.
(1991) Chapter in Peptides as Probes in Muscle Research,
Springer-Verlag, p.57-68. Ed: Johann Caspar Ruegg
The use of peptide mimetics to define the actin binding sites on
the head of the myosin molecule.
|
|---|
|
Synopsis:
Protein-protein interactions are involved in muscle contraction and
signal transduction. This book describes how synthetic peptides may be
used, much like antibodies, both as specific inhibitors and as molecular
probes to explore the cognitive interfaces between interacting proteins
and their functional significance. This offers the prospect of very
selective intervention in cellular mechanisms. These contributions by
several experts should appeal to the researchers in muscle physiology,
cardiovascular pharmacology and cell biology who are interested in this
new approach.
ISBN: 3540536531 and ASIN: 0387536531
Still available from
Amazon.
|
|
Alessi,D.R. Smith,K.J., & Trayer,I.P.
(1992) J.Muscle Res.Cell Motil., 13, 220.
Short Report: Location of an S1 binding site on an
a-helical region of actin.
|
|---|
|
Abstract:
An 18-residue peptide from actin comprising of residues 77-95
(T77NWDDMEKIWHHTFYNEL94) has been
synthesized and purified. According to the recent published actin
structure (Kabsch, W. et al. (1990) Nature 347, 37-44), residues 79-92
form a surface a-helix at the top of subdomain 1.
In aqueous solution at pH 7.8, and over a temperature range
10-26oC, extensive two-dimensional
NMR studies have shown that this peptide adopts an a-helical structure from
W79-H88. At this point the helix is
disrupted but appears to reform towards the C-terminus. Even in the
helix-inducing solvent, trifluoroethanol, where the helical regions are
strengthened, this helix break is still found. NMR studies have also shown
that this peptide binds to S1, predominantly around the helix disruption
region and at selected other amino acids. The specificity of this binding
is shown by the fact that the peptide is readily displaced from S1 by
actin and that the interaction still occurs when the ionic strength is
raised to 0.5 M. After iodination with
125I, the peptide was crosslinked to S1 with the
zero length crosslinker EDC. Radioactivity was only associated with the
tryptic 50 kDa domain and has so far been located in the N-terminal 40 kDa
fragment of this domain. The exact interaction site is currently under
investigation. Rather interestingly, this peptide does not appear to
inhibit or activate the actin-stimulated Mg.ATPase of S1. We have been
using this peptide to probe the architecture of S1 by using the distance
dependent broadening effect of suitably located nitroxide spin labels on
the peptide 1H-NMR signals. A spin-labelled ADP
analogue (Alessi, D. et al. (1991) J. Chem. Soc. ) in the active site of
S1 was found to broaden resonances from the peptide bound to S1 indicating
that the two sites are < 1.5 nm apart. The spin-labeled
ATP has been shown to be hydrolyzed by S1 and to support muscle
contraction). Furthermore, a nitroxide spin label attached to
707Cys, situated in another actin-binding site
on S1, also broadens resonances arising from this actin peptide,
emphasizing the spatial proximity of the two actin-binding sites on
different domains of S1. Reduction of both spin-labeled moieties in situ
with ascorbate removes their paramagnetic effects. Comparison of the
binding of this peptide to S1 and to an S1.ATP analogue state (pPDM-S1),
the latter representing a weak-binding S1.ATP conformation, reveals that
the actin peptide binds differently and more weakly to the pPDM-S1. That
is, this actin-binding site can distinguish between the strong- and
weak-binding S1 states.
Local backup of Full text webpage
and here is the full article pdf.
get this article from the
Journal of Muscle Research and Cell Motility
site
|
|
Smith,K.J.
(1993) PhD. Thesis, Dept. Biochemistry, University of Birmingham.
The Use of Peptide Mimentics to Investigate the interface between Actin and Myosin.
|
|---|
|
Abstract:
It has been known for some time that small peptides corresponding to
certain sequences in proteins are capable of adopting conformations in
aqueous solution that are similar to the conformation in the native
protein (eg. Goodman & Kim, 1989, Biochemistry, 28, 4343-4347). Small
peptides corresponding to sequences in actin and myosin, which are thought
to be interaction sites between actin and myosin, are known to interact
with the reciprocal protein partner in such a way that they interfere with
the biological activity of the actomyosin system in assays both using the
isolated muscle proteins (eg. inhibition of the actin activated
Mg2+ATPase activity) and using intact myofibrils
(eg. inhibition of force generation; eg. Keane et al., 1990, Nature 344,
265-268). The relationship between the structure adopted by these peptides
and the biological activity has not previously been investigated. In this
thesis peptides from both actin and myosin have been identified which
specifically bind to the protein partner (myosin S1 peptides corresponding
to sequences from the 20 kDa domain 704-710 and 718-727, and from 50 kDa
domain 609-618, each of which bind to F-actin; actin peptides: 16-41,
77-94, 29-58 and 96-117, the first two of which bind to the myosin S1
head). The binding residues within these peptides have been identified by
the observation of broadening of resonances in NMR and, for some of the
peptides, the site of interaction in the protein template has been
determined by chemically crosslinking the radioactively labelled peptide
to the protein. In addition for many of the peptides identified as binding
sites, the solution structure of the isolated peptide was determined by
NMR spectroscopy (and in one case by CD spectroscopy). In particular a
helical peptide (actin 77-94) and extended peptide (myosin 718-727) were
investigated. In each case an attempt was made to relate the solution
structure to the observed binding properties. In the two peptides
corresponding to actin 77-95 and myosin 718-727, transferred NOESY spectra
were recorded in the presence of the protein partner, and some
observations of the conformation adopted by the peptide when bound to the
protein template were made. In addition the spatial relationship between
the SH1 (707Cys) and SH2
(697Cys) thiols and the ATPase site in the S1
head was investigated by 19F labelling the thiol
and using a spin labelled analogue of ATP. A region of the myosin S1 head
corresponding to residues 601-635 was sequenced.
A preliminary HTML version of my thesis available -
here (no figures yet).
See the thesis collection catalogue at the
Birmingham University library
and search for
"Smith, Kevin John. - The use of peptide
mimetics to investigate the interface between actin and m. - Birmingham :
University of Birmingham, 1992. - v8157600 "
or get it from the Main
Library at Thesis Store Diss.S2.B93
|
|
Smith,K.J., Eastwood,A.M. and Trayer,I.P.
(1993) J.Muscle Res. Cell Motil., 14, 272.
Short Report: Analysis of the weak binding site on actin.
|
|---|
|
Abstract:
We recently reported that an 18-residue peptide, comprising residues
77-95 of the actin structure, forms part of the myosin binding site on
actin (Alessi et al. (1992) J. Muscle Res. Cell Motil. 13, 220). In actin,
this region is largely a-helical, with an
unwinding of the helix at the C-terminus (a-helix
-> 5-turn helix), and is located at the top
and 'side' of subdomain 1. This peptide binds equally well to S1 alone and
to an S1-ATP analogue state, suggesting it forms a contact point in the
weak binding acto-S1-ATP complex which is consistent with recent image
reconstruction studies (Milligan and Holmes, unpublished data). Extensive
analysis of this peptide in solution and when bound to S1 has been carried
out by NMR and its structure determined from NOESY spectra using the
simulated annealing protocol of X-PLOR. In 50% aqueous trifluoroethanol
(TFE), the peptide was essentially a-helical over
most of its length with both the backbone and side chains aligned. The
helix was slightly curved and further analysis showed that there was a
discontinuity between H87 and
H88 that was most apparent in the Ramachandran
plots and seemed to originate at a break in the regular organization of
the side chains; up to H87 the side chains were
aligned C->N and after H88 they
were aligned N->C. In most a-helices
the side chains align either
N->C or C->N without the break. In 90%
H2O, the helical character of the backbone is
still apparent although the side chains are more flexible and the
C-terminal end unwinds out of the a-helix, i.e.
in some respect it is more like the structure found in actin. Using the
transferred NOESY method, the structure of the peptide bound to S1 in 90%
H2O was also determined. This was found to be
much more like that in 50% TFE as evidenced by the larger number of
transferred NOEs than NOEs found in water in the absence of S1. The
backbone was mostly helical and the side chains more ordered. In
particular, residues W86-
H87, were held relatively rigid indicating that
these two amino acids were the major contact points with S1. In actin,
H87 (and H88) are
readily available for interaction with S1 but
W86, is buried. However, limited molecular
dynamics calculations based on minor changes in winding of the C-terminal
region of this helix show that W86, can be
exposed (and H88 buried) at this discontinuity
point in the helix. This could be a means whereby S1 could deform the
actin structure and transmit information through the molecule. As controls
it has shown that peptides containing the a-
helical region 113-125 also retain their structure in
solution but do not bind to S1. This is the major surface feature of the
'back' of the subdomain 1. We are presently investigating peptides
containing the 338-348 helix, which is a dominant feature of the 'front'
of subdomain 1.
Local backup of Full text PDF
and here is the full article pdf.
get this article from the
Journal of Muscle
Research and Cell Motility site
|
|
Jaseja,M., Smith,K.J., Lu,X., Williams,J.A., Trayer,H.R.,
Trayer,I.P. and Hyde,E.I.
(1993) Eur. J. Biochem., 218, 853-860.
1H-NMR studies and the secondary structure of the
RGD-containing snake toxin, albolabrin.
|
|---|

Abstract:
Albolabrin is a naturally occurring peptide from snake venom containing
the sequence Arg-Gly-Asp (RGD). It inhibits platelet aggregation by
blocking the binding of fibrinogen to the glycoprotein Gp IIb-IIIa, on the
surface of activated platelets. Albolabrin consists of 73 residues with
six intramolecular disulphide bonds. The 1H-NMR
spectrum of albolabrin has been assigned using homonuclear two-dimensional
techniques and its secondary structure determined. Like kistrin and
echistatin, two related peptides from snake venom, albolabrin appears to
have little regular secondary structure in solution. Several bends and two
short distorted beta sheets are observed. The RGD sequence, important for
binding to the receptor, lies in a mobile loop joining two strands of one
of these beta sheets. This loop undergoes a pH-dependent conformational
change.
Local backup of Full text webpage
and here is the full article pdf.
get this article from the
European Journal of Biochemistry site (Now the FEBS Journal).
|
|
Smith,K.J., Trayer,I.P. and Grand, R.J.A.
(1994) Biochemistry, 33, 20, 6063-6073.
Structure around the cleavage site in the thrombin receptor
determined by NMR spectroscopy.
|
|---|
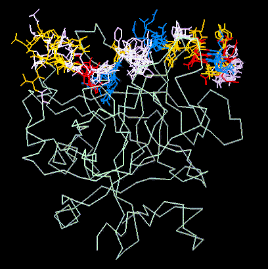
Abstract:
NMR spectroscopic experiments were performed to study the structure of
synthetic peptides identical to two extracellular regions of the human
thrombin receptor. The smaller molecule, comprising 14 amino acids, was
biologically active and was equivalent to the ''tethered ligand'' exposed
after cleavage of the receptor by thrombin. The principal structural
elements were two overlapping turns (amino acids 5-8 and 6-9), the second
of which was stabilized by a hydrogen bond,
6(CO)- 9(NH). The five
N-terminal residues, considered to be responsible for biological activity,
were essentially unstructured. A second version of this peptide,
biologically inactive due to the reversal of the two N-terminal amino
acids, had a very similar structure. A longer peptide (23 amino acids)
covering the proposed thrombin cleavage site was found to be more highly
structured. The seven residues from Pro(-2) to
Arg(5) (the N-terminal amino acid exposed after
cleavage is taken as residue 1) formed a 3(10)
helix which is not present in the shorter tethered ligand peptide. The
structure is partially stabilized by a charged hydrogen bond between the
side chains of Arg(-1) and
Asp(-3). The overlapping turns observed in the
shorter peptides could also be distinguished in the longer molecule. On
the basis of the structure determined for the peptide which encompasses
the cleavage site and the determined structure of thrombin, a model is
postulated for the interaction of the thrombin receptor and the protease
during activation.
Local backup of Full text webpages
and here is the full article pdf.
get this article from the
Biochemistry site
|
|
Trayer,I.P, Smith,K.J., Eastwood,A.M., and Trayer,H.R.
(1995) J.Musc.Res.Cell.Motil., 16, 450.
Short Report: Peptide mimetics as a probe of the contractile cycle.
|
|---|
|
Abstract:
The various sites by which actin and myosin contact each other during
the contractile cycle have been identified using synthesized peptides to
mimic each individual site in turn and employing the partner protein to
act as the molecular template. Specific binding has been established by
using NMR techniques, by showing they were competitive inhibitors of the
acto-S1 Mg.ATPase activity and by direct binding studies. The structure of
each peptide in solution has been investigated by 2D-NMR techniques and
was found to possess the same elements of secondary structure as in the
parent protein. In some cases, the structure of the peptide bound to its
partner has been determined by the transferred NOESY experiment which
revealed subtle changes in the peptide structure on binding, suggesting
how the allosteric response between different sites might be communicated.
The sites on actin that have been identified as binding to S1 are
contained within the N-terminal region, residues 338-349, 77-94 and the
loop around residue 40. The sites on S1 identified as containing residues
binding to actin are 400-416, 528-552, and 568-579. Experiments monitoring
the displacement of S1 from actin (± ATP) have allowed contacts that occur
in the weakly attached acto-S1 complex to be distinguished from the
strongly attached states and have suggested an ordered and sequential
docking of the two proteins.
Local backup of Full text PDF
and here is the full article pdf.
get this article from the
Journal of Muscle
Research and Cell Motility site
|
|
Smith,K.J., Jaseja,M., Lu,X., Williams,J.A., Trayer,I.P.,
Hyde,E.I. and Trayer,I.P.
(1995) Intl. J. Peptide & Protein Research, 48, 3,
220-228.
3-Dimensional structure of the RGD-containing snake toxin
Albolabrin in solution, based on 1H-NMR spectroscopy and
simulated annealing calculations.
|
|---|
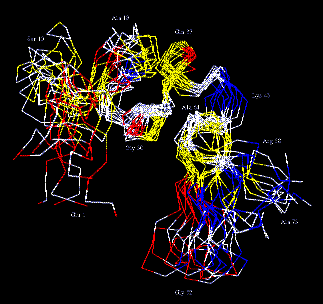
Abstract:
Albolabrin is a snake toxin that contains a RGD-(Arg-Gly-Asp) sequence
motif and competes with fibrinogen to bind to the integrin alpha IIb beta
3 (GpIIb-IIIa) on platelets. It thus inhibits platelet aggregation and
cell-cell adhesion. It shows a high sequence similarity to other
disintegrins, yet the reported disulfide bonding pattern for this peptide
differs from that of others in this family. Recently we reported the
assignment of the 1H-NMR spectrum of albolabrin
and a preliminary description of its secondary structure [Jaseja, M.,
Smith, K.J., Lu, X., Williams, J.A., Trayer, H., Trayer, I.P. & Hyde,
E.I. (1993) Eur. J. Biochem. 218, 853-860]. Here we present a more
detailed description of the secondary and the tertiary structure, based on
the 1H NMR results and simulated annealing
methods. The structure of albolabrin in solution was calculated using 318
distance and 18 dihedral angle restraints. The average atomic RMS
deviation between 12 refined structures and the mean structure was 3.1
Angstrom for the backbone. The protein appears to be highly mobile. Its
structure is dominated by a series of turns and by three hairpins, each
with a short region of distorted antiparallel beta- pleated sheet, held
together by six disulfide bridges. The most well defined area is the
hydrophobic core, residues 21-37 and 57-67, which is clustered around F40
and has a backbone atomic RMS deviation of only 1.3 Angstrom from the mean
structure. The RGD adhesion sequence is found at the highly mobile tip of
one of the beta-hairpins, protruding from the body of the protein. Many of
these structural features are similar to those of other disintegrins, and
differences in the disulfide bonding pattern of the disintegrins can be
accomodated without significant energy penalty. Comparison of this
structure with other proteins of similar function suggests that it is the
RGD-loop, rather than the precise topology of the proteins, that is
important to antagonist activity.
Sketch of the calculated structure of albolabrin
PDB file not available.
Local backup of Full text webpage
and here
or here is the full article pdf.
the International Journal of Peptide and Protein research and Peptide
Research was succeded by the Journal of Peptide Research, which is now defunct.
|
|
Smith,K.J. , Scotland,G. Beattie,J. Trayer,I.P. and Houslay,M.D.
(1996) J.Biol.Chem., 271, 28, 16703-16711.
Determination of the structure of the N-terminal splice region of
the cyclic AMP-specific phosphodiesterase RD1 (RNPDE4A1) by
1H-NMR and identification of the membrane
association domain using chimeric constructs.
|
|---|
|
Abstract:
A 25-residue peptide representing the membrane targeting N-terminal
splice region of the cyclic AMP phosphodiesterase RD1 (RNPDE4A1) was
synthesized, and its structure was determined by
1H-NMR, Two independently folding helical
regions were identified, separated by a highly mobile ''hinge'' region,
The first helical region was formed by an N-terminal amphipathic
a-helix, and the second consisted of multiple
overlapping turns and contained a distinct compact, hydrophobic,
tryptophan-rich domain (residues 14-20), Chimeric molecules, formed
between the N-terminal region of RD1 and the soluble bacterial protein
chloramphenicol acetyltransferase, were used in an in vitro system to
determine the features within the splice region that were required for
membrane association, The ability of RD1-chloramphenicol acetyltransferase
chimera to become membrane-associated was not affected by deletion of any
of the following regions: the apolar section (residues 2-7) of the first
helical region, the polar part of this region together with the hinge
region (residues 8-13), or the polar end of the C-terminal helical region
(residues 21-25), In marked contrast, deletion of the compact, hydrophobic
tryptophan-rich domain (residues 14-20) found in the second helical region
obliterated membrane association, Replacement of this domain with a
hydrophobic cassette of seven alanine residues also abolished membrane
association, indicating that membrane association occurred by virtue of
specific hydrophobic interactions with residues within the compact,
tryptophan-rich domain, The structure of this domain is well defined in
the peptide, and although the region is helical, both the backbone and the
distribution of side chains are somewhat distorted as compared with an
ideal a-helix, Hydrophobic interactions, such as
the ''stacked'' rings of residues Pro(14) and
Trp(15), stabilize this domain with the side
chain of residue Leu(16) adopting a central
position, interacting with the side chains of all three tryptophan
residues 15, 19, and 20, These bulky side chains thus form a hydrophobic
cluster, In contrast, the side chain of residue
Val(17) is relatively exposed, pointing out from
the opposite ''face'' of the peptide, Although it appears that from the
opposite ''face'' of the peptide, Although it appears that this compact,
tryptophan-rich domain is responsible for membrane association, at present
the target site and hence the specific interactions involved in membrane
targeting by the RD1 splice region remain unidentified.
|
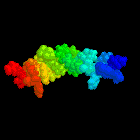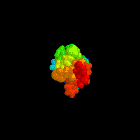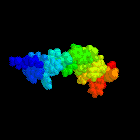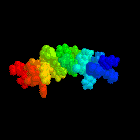 Local backup of Full text PDF
and here is the full article pdf.
get this article from the
JBC
site
See also Brookhaven
Database PDB entry 1LOI.
launch your molecular graphics software with 1loi.pdb
the pdb coordinates entry for 1loi.pdb
the pdb restraints entry for 1loi.pdb
Local backup of Full text PDF
and here is the full article pdf.
get this article from the
JBC
site
See also Brookhaven
Database PDB entry 1LOI.
launch your molecular graphics software with 1loi.pdb
the pdb coordinates entry for 1loi.pdb
the pdb restraints entry for 1loi.pdb
|

An illustration of this structure was used as the cover for the
Biochemical Journal during the whole of 1997.
The Biochemical Journal site
large version of the cover.
M. Houslay, J. Smith, G.Scotland, J. Beattie, I. Trayer.
Studies on the RD1 peptide with
Prof. Miles Houslay, at Glasgow Univ.
|
|
I.P.Trayer and Smith,K.J.
(1997) Trends in Cell Biology, 7, 7, 259-263.
Review: Motoring down the highways of the cell.
|
|---|
|
Abstract:
All eukaryotic cells contain large numbers of motor proteins (kinesins,
dyneins and myosins), each of which appears to carry out aspecialized
force-generating function within the cell. They are known to have roles in
muscle contraction, ciliary movement, organelle andvesicle transport,
mitosis and cytokinesis. These motor proteins operate on different
cytoskeletal filaments; myosins move along actin filaments, and kinesins
and dyneins along microtubules. Recently published crystal structures of
the motor domains of two members of the kinesin superfamily reveal that
they share the same overall fold the kinesin superfamily reveal that they
share the same overall fold that is also found at the core of the larger
myosin motor. This suggests that they may share a common mechanism as well
as a common ancestry.
pdf format figure 1
pdf format figure 3
Local backup of Full text webpage
and here is the full article pdf,
and here is the full color article pdf.
get this article from the
Trends in Cell Biology site
|
|
Jeeves,M. Smith,K.J. Quirk,P.G. Cotton,N.P.J. & Jackson,J.B.
(1999) J.Biomol.NMR 13, 305-306.
Assignment of the NADP(H)-binding Component (domain III) of the
proton-translocating transhydrogenase from Rhodospirillum rubrum.
|
|---|
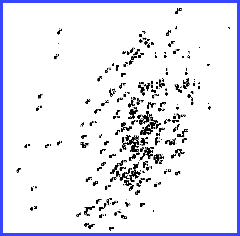
The 1H,
15 N spectrum of domain III.
Local backup of Full text PDF
and here is the full article pdf.
get this article from the
Journal of Biomolecular NMR site
larger annotated spectrum
full assignments text
See also our BMRB entry (4236)
http://www.bmrb.wisc.edu/
|
|
Molloy,D.P. Smith,K.J. Milner,A.E. Gallimore,P.H. & Grand,R.J.A.
(1999) J. Biol. Chem., 274, 6, 3503-3512.
The Structure of the Site on Adenovirus Early Region 1A Responsible for
Binding to TATA-binding Protein Determined by NMR Spectroscopy.
|
|---|
|
Abstract:
Previous detailed mutational analysis has shown that the binding site
on adenovirus (Ad) early region 1A (E1A) for TATA-binding protein (TBP) is
located toward the N terminus of conserved region 3 (CR3). Here we
demonstrate that synthetic peptides of between 15 and 22 amino acids,
identical to amino acid sequences of CR3 present in the larger Ad5 E1A (13
S product) and in both the Ad12 E1A (13 and 12 S products) proteins that
lie N-terminal to the zinc finger motif, can disrupt binding of E1A to
TBP. These findings suggest that the peptides are biologically active in
terms of interacting with TBP and must therefore comprise some, if not
all, of the TBP binding site on E1A. The interaction between Ad12 E1A and
TBP was confirmed by direct co-precipitation experiments. In
1H NMR studies of CR3 peptides, regular patterns
of NOEs were observed from which their conformational preferences in
aqueous solution were determined. Both Ad5 and Ad12 peptides were shown to
contain regions of helical backbone structure in 50% trifluoroethanol. In
each case, the type and intensities of NOE cross-peaks observed correlated
best to a-helical turns. These helices are more
extensive in larger peptides and extend from
Glu141 to Val147 and
from Arg144 to Pro152
in the full-length Ad5 and Ad12 13S E1A proteins, respectively. The
structure of a 19-residue Ad5 CR3 peptide carrying the V147L mutation in
the full-length protein that abolishes TBP binding was examined. No
significant differences between the substituted and wild type peptides
were observed, suggesting that this substitution in the intact protein may
cause disruption of global rather than local structures.
Local backup of Full text PDF
and here is the full article pdf.
get this article from the
JBC
site
|
|
Timson,D.J. Trayer,H.R. Smith,K.J. & Trayer,I.P.
(1999) J.Biol.Chem. 274(26), 18271-18277
Size and Charge Requirements for Kinetic Modulation and Actin
Binding by A1-type Myosin Essential Light Chains.
|
|---|
|
Abstract:
There are two forms of essential light chain (ELC) in vertebrate
striated muscle: A1-type and A2-type. Consequently there are two
isoenzymes of myosin subfragment 1 prepared by chymotryptic digestion of
myosin: S1A1 and S1A2 depending on which ELC is associated with the heavy
chain. A1-type ELCs have an additional 40 or so amino acids residues at
the N-terminus compared to the A2-type. These additional amino acids in
A1-type ELCs are responsible for binding actin and modulating the kinetic
activity of myosin molecules that carry them. The requirements for actin
binding and kinetic modulation of the myosin motor by an A1-type ELC have
been investigated by a protein engineering approach. Mutations in the
actin binding site of the human atrial ELC (HmAtELC) result in altered
actin activated MgATPase kinetics when the recombinant light chains are
hybridised into rabbit skeletal myosin subfragment 1. Substitution of the
positively charged lysine residue at position 4 with the neutral amino
acid alanine results in decreased kinetic modulation (the resulting hybrid
S1 has kinetic parameters approximately midway between S1A1 and S1A2 under
the same conditions) and decreased actin binding (as judged by chemical
cross-linking). Replacement of this residue with aspartate (negatively
charged) reduces actin binding still further and the hybrid S1 is almost
S1A2-like in its kinetic parameters. Substitution of lysine 3 by alanine
gives similar results to same replacement at position 4. However, the size
of the side chain at position 1 is unimportant: alteration of the wild
type amino acid (alanine) to valine results in no change in the kinetic
parameters of the hybrid S1 or in the ELC's ability to bind actin.
Furthermore we found a statistically significant positive correlation
between the apparent KM for actin and the
Kcat for Mg.ATP turnover for each mutant hybrid.
This strengthens our belief that the binding of actin by A1-type ELCs
results directly in modulation of the myosin motor.
Local backup of Full text PDF
and here is the full article pdf.
see the
JBC
site
|
|
Quirk,P.G. Smith,K.J.. Thomas, C. & Jackson,J.B.
(1999) Biochimica et Biophysica Acta (BBA)/Bioenergetics
1412(2), 139-148
The Mobile Loop Region of the NAD(H)-binding component (dI) of
proton-translocating Nicotinamide Nucleotide Transhydrogenase from
Rhodospirillum rubrum: Complete NMR assignment and effects of bound
nucleotides.
|
|---|
|
Abstract:
The dI component of transhydrogenase binds NAD+ and NADH. A mobile loop
region of dI plays an important role in the nucleotide binding process,
and mutations in this region result in impaired hydride transfer in the
complete enzyme. We have previously employed one-dimensional 1H-NMR
spectroscopy to study wild-type and mutant dI proteins of Rhodospirillum
rubrum and the effects of nucleotide binding. Here, we utilise two- and
three-dimensional NMR experiments to assign the signals from virtually all
of the backbone and side-chain protons of the loop residues. The mobile
loop region encompasses 17 residues: Asp223-Met239. The assignments also
provide a much strengthened basis for interpreting the structural changes
occurring upon nucleotide binding, when the loop closes down onto the
surface of the protein and loses mobility. The role of the mobile loop
region in catalysis is discussed with particular reference to a
newly-developed model of the dI protein, based on its homology with
alanine dehydrogenase.
Local backup of Full text PDF
and here is the full article pdf.
get this article from the
BBA-Bioenergetics site
|
|
Philip G. Quirk, Mark Jeeves, Nick P.J. Cotton, John K. Smith
& Baz J. Jackson
(1999) FEBS Letters, 446, 1, 127-132.
Structural changes in the recombinant, NADP(H)-binding component
of proton translocating transhydrogenase revealed by NMR spectroscopy
|
|
Abstract:
We have analysed 1H,
15N-HSQC spectra of the recombinant,
NADP(H)-binding component of transhydrogenase in the context of the
emerging three dimensional structure of the protein. Chemical shift
perturbations of amino acid residues following replacement of
NADP+ with NADPH were observed in both the
adenosine and nicotinamide parts of the dinucleotide binding site and in a
region which straddles the protein. These observations reflect the
structural changes resulting from hydride transfer. The interactions
between the recombinant, NADP(H)-binding component and its partner,
NAD(H)-binding protein, are complicated. Helix B of the recombinant,
NADP(H)-binding component may play an important role in the binding
process.
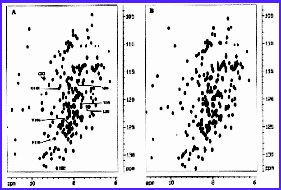 The 1H,
15N spectrum of domain III bound to
NADP+ and NADPH.
The 1H,
15N spectrum of domain III bound to
NADP+ and NADPH.
Local backup of Full text
PDF
and here is the full article pdf.
get this article from the
FEBS site
|
|
Jeeves M.; Smith K.J.; Quirk P.G.; Cotton N.P.J.; Jackson J.B.
(2000) Biochimica et Biophysica Acta (BBA)/Bioenergetics,
1459(2), 248-257
Solution structure of the NADP(H)-binding component (dIII) of
proton-translocating transhydrogenase from Rhodospirillum rubrum.
|
|---|
|
Abstract:
Transhydrogenase is a proton pump found in the membranes of bacteria
and animal mitochondria. The solution structure of the expressed, 21.5
kDa, NADP(H)-binding component (dIII) of transhydrogenase from
Rhodospirillum rubrum has been solved by NMR methods. This is the first
description of the structure of dIII from a bacterial source. The protein
adopts a Rossmann fold: an open, twisted, parallel -sheet, flanked by
helices. However, the binding of NADP+ to dIII is profoundly different to
that seen in other Rossmann structures, in that its orientation is
reversed: the adenosine moiety interacts with the first motif, and the
nicotinamide with the second. Features in the structure that might be
responsible for changes in nucleotide-binding affinity during catalysis,
and for interaction with other components of the enzyme, are identified.
The results are compared with the recently determined, high-resolution
crystal structures of human and bovine dIII which also show the reversed
nucleotide orientation.
Local backup of Full text PDF
and here is the full article pdf.
get this article from the
BBA-Bioenergetics site
|
|
J. W. Emsley,D. Merlet, K. J. Smith, and N. Suryaprakash
(2002) Journal of Magnetic Resonance 154, 303-310
Selective Detection of the Proton NMR Spectra of Molecules
Containing Rare Spins at Natural Abundance in Liquid Crystalline Samples
|
|---|
|
Abstract:
It is shown that the proton NMR spectra of molecules containing rare
spins at natural abundance dissolved in a liquid crystalline solvent can
be obtained free from the strong lines from the spectrum of the abundant
isotopomer by the 2D HSQC NMR experiment. The technique can also give the
individual chemical shifts of the rare spins, and, for a molecule
containing another abundant nucleus, such as fluorine, the rare spin
19F total anisotropic couplings are also
obtained. The usefulness of the technique is demonstrated for molecules
containing 13C as the rare spins.
Local backup of Full text PDF
and here is the full article pdf.
or from the
Journal
of Magnetic Resonance site
|
|
Ray P., Smith K.J., Parslow R.A., Dixon R., Hyde E.I.
(2002) Nucleic Acids Research 30(18), 3972-3980
Secondary structure and DNA binding by the C-terminal domain of
the transcriptional activator NifA from Klebsiella pneumoniae
|
|---|
|
Abstract:
The NifA protein of Klebsiella pneumoniae is required for
transcriptional activation of all nitrogen fixation (nif ) operons except
the regulatory nifLA genes. At these operons, NifA binds to an upstream
activator sequence (UAS), with the consensus TGTN10-ACA, via a C-terminal
DNA-binding domain (CTD). Binding of the activator to this upstream
enhancer-like sequence allows NifA to interact with RNA polymerase
containing the alternative sigma factor, sigma54. The isolated NifA CTD is
monomeric and binds specifically to DNA in vitro as shown by DNase I
footprinting. Heteronuclear 3D NMR experiments have been used to assign
the signals from the protein backbone. Three a-helices have been
identified, based on secondary chemical shifts and medium range Hai-NHi +
1, and NHi-NHi + 1 NOEs. On addition of DNA containing a half-site UAS,
several changes are observed in the NMR spectra, allowing the
identification of residues that are most likely to interact with DNA.
These occur in the final two helices of the protein, directly confirming
that DNA binding is mediated by a helix±turn±helix motif.
Local backup of Full text PDF
and here is the full article pdf,
and here is the supplementary material.
get this article from the
Nucleic
Acids Research site
|
|
K John Smith, George S Baillie, Eva I Hyde, Xiang Li,
Thomas M Houslay, Angela McCahill, Allan J Dunlop,
Graeme B Bolger, Enno Klussmann, David R Adams, Miles D Houslay
(2007) Cellular Signalling. 19(12), 2612-2624
(1)H NMR structural and functional characterisation of a cAMP-specific
phosphodiesterase-4D5 (PDE4D5) N-terminal region peptide that disrupts PDE4D5
interaction with the signalling scaffold proteins, betaarrestin and RACK1.
|
|---|
|
Abstract:
The unique 88 amino acid N-terminal region of cAMP-specific phosphodiesterase-4D5 (PDE4D5) contains overlapping binding sites
conferring interaction with the signaling scaffold proteins, betaarrestin and RACK1. A 38-mer peptide,
whose sequence reflected residues 12 through 49 of PDE4D5, encompasses the entire N-terminal RACK1 Interaction Domain
(RAID1) together with a portion of the betaarrestin binding site. (1)H NMR and CD analyses indicate that this region has
propensity to form a helical structure. The leucine-rich hydrophobic grouping essential for RACK1 interaction forms a discrete
hydrophobic ridge located along a single face of an amphipathic alpha-helix with Arg34 and Asn36, which also play important roles
in RACK1 binding. The Asn22/Pro23/Trp24/Asn26 grouping, essential for RACK1 interaction, was located at the N-terminal head of the
amphipathic helix that contained the hydrophobic ridge. RAID1 is thus provided by a distinct amphipathic helical structure. We
suggest that the binding of PDE4D5 to the WD-repeat protein, RACK1, may occur in a manner akin to the helix-helix interaction shown
for G(gamma) binding to the WD-repeat protein, G(beta). A more extensive section of the PDE4D5 N-terminal sequence (Thr11-Ala85) is
involved in betaarrestin binding. Several residues within the RAID1 helix contribute to this interaction however. We show here that
these residues form a focused band around the centre of the RAID1 helix, generating a hydrophobic patch (from Leu29, Val30 and Leu33)
flanked by polar/charged residues (Asn26, Glu27, Asp28, Arg34). The interaction with betaarrestin exploits a greater circumference on
the RAID1 helix, and involves two residues (Glu27, Asp28) that do not contribute to RACK1 binding. In contrast, the interaction of
RACK1 with RAID1 is extended over a greater length of the helix and includes Leu37/Leu38, which do not contribute to betaarrestin
binding. A membrane-permeable, stearoylated Val12-Ser49 38-mer peptide disrupted the interaction of both betaarrestin and RACK1 with
endogenous PDE4D5 in HEKB2 cells, whilst a cognate peptide with a Glu27Ala substitution selectively failed to disrupt PDE4D5/RACK1
interaction. The stearoylated Val12-Ser49 38-mer peptide enhanced the isoprenaline-stimulated PKA phosphorylation of the
beta(2)-adrenergic receptors (beta(2)AR) and its activation of ERK, whilst the Glu27Ala peptide was ineffective in both these regards.
Local backup of Full text PDF
and here is the full article pdf.
get this article from the
Cellular Signalling site
|
|
|
Full set of configuration file for Irix6.5
from our server.
Links to my Varian pulse programs, macros, calibrations, etc...
Full text of grants ,
link to my conference posters ,
and slides/overheads.
|
|
|

{kind=link}